Total workflow
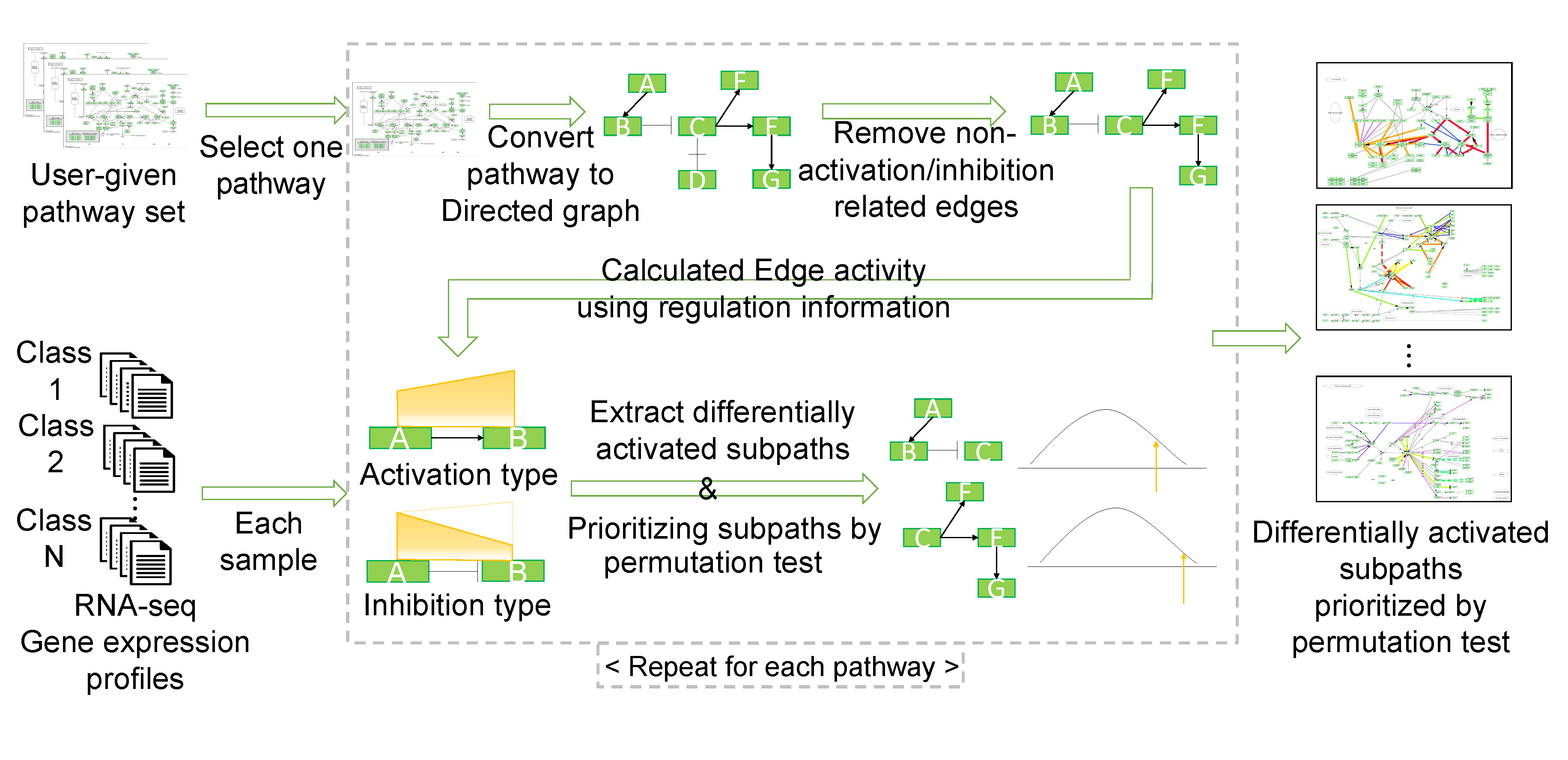
MIDAS: MIning Differentially Activated Subpaths of KEGG pathways from multi-class RNA-seq data
Abstract
Pathway based analysis of high throughput transcriptome data is a widely used approach to investigate biological mechanisms. Since a pathway consists of multiple functions, the recent approach is to determine condition specific sub-pathways or subpaths. However, there are several challenges. First, few existing methods utilize explicit gene expression information from RNA-seq. More importantly, subpath activity is usually an average of statistical scores, e.g., correlations, of edges in a candidate subpath, which fails to reflect gene expression quantity information. In addition, none of existing methods can handle multiple phenotypes.
To address these technical problems, we designed and implemented an algorithm, MIDAS, that determines condition specific subpaths, each of which has different activities across multiple phenotypes. MIDAS utilizes gene expression quantity information fully and the network centrality information to determine condition specific subpaths. To test performance of our tool, we used TCGA breast cancer RNA-seq gene expression profiles with five molecular subtypes. 36 differentially activate subpaths were determined.
The utility of our method, MIDAS, was demonstrated in four ways. All 36 subpaths are well supported by the literature information. Subsequently, we showed that these subpaths had a good discriminant power for five cancer subtype classification and also had a prognostic power in terms of survival analysis. Finally, in a performance comparison of MIDAS to a recent subpath prediction method, PATHOME, our method identified more subpaths and much more genes that are well supported by the literature information.
Highlights of MIDAS
- MIDAS utilizes explicit gene expression quantity information from RNA-seq.
- MIDAS extends a recent edge activation measurement technique for determining subpaths with differential activities.
- MIDAS uses the multi-class issue in a statistical approach.
- MIDAS uses a greedy subpath extension method with exponentially increasing criteria.
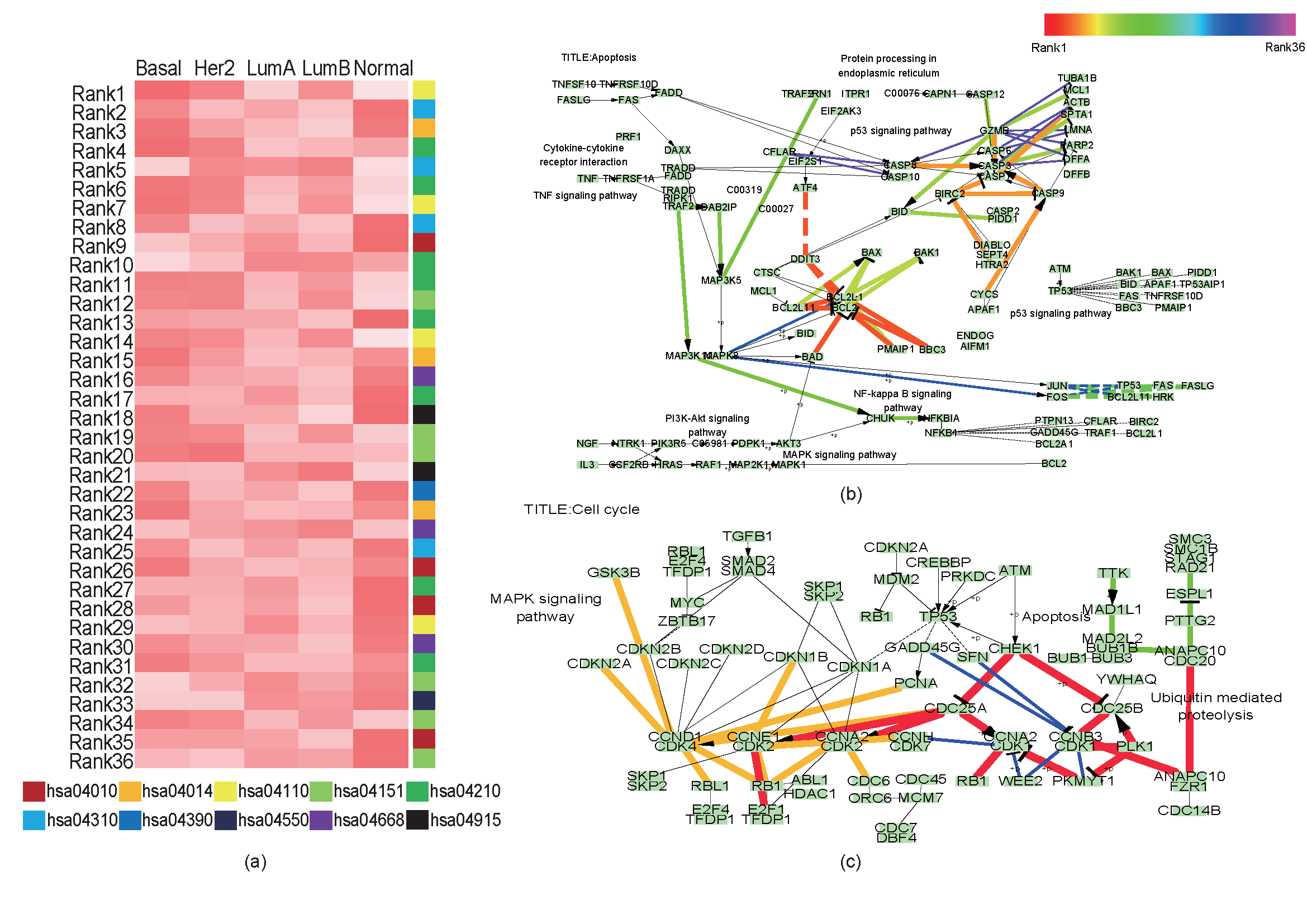
Average subpath activity among breast cancer subtypes and Subpaths result: (a) average subpath activity is coded as color heatmap. Red color denotes higher subpath activity and white denotes lower subpath activity. (b) and (c) are results where differentially activated subpaths are located. Those subpaths are decoded as rainbow color scheme and edge widths accoriding to their rank. The higher rank subpath is more thicker and red side color. (b) is result of Apoptosis (hsa04210). (c) is result of Cell cycle (hsa04110).