Model Description
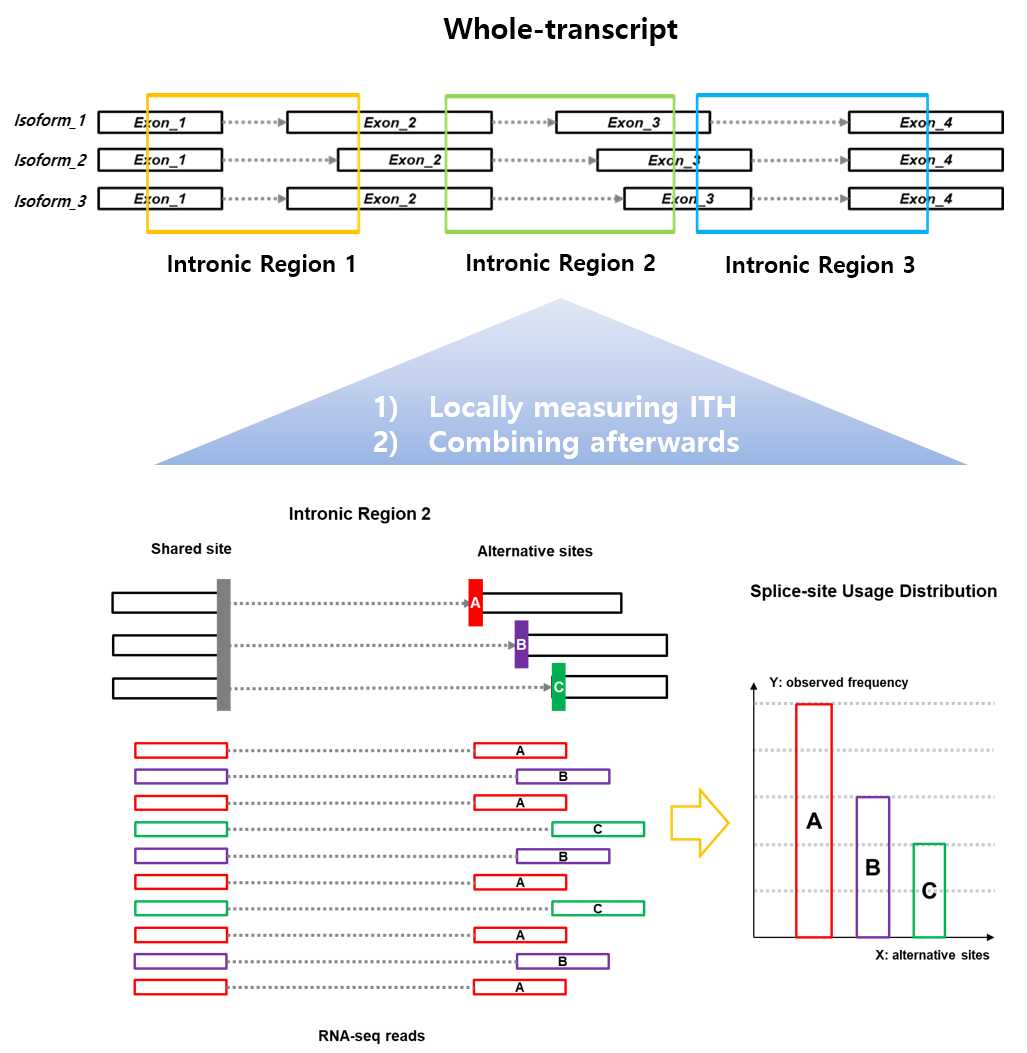
Figure 1. Illustration for an intronic splicing unit. Splicing unit is defined as a collection of splicing events sharing a common splice-site (i.e. donor or acceptor) in an intronic region. Splicing unit consists of the splice-site usage distribution of each sample at each locus. Here, the splice-site usage distribution is calculated by the number of RNA-seq reads supporting each alternative splice-site (colored red, purple, and green in the figure).
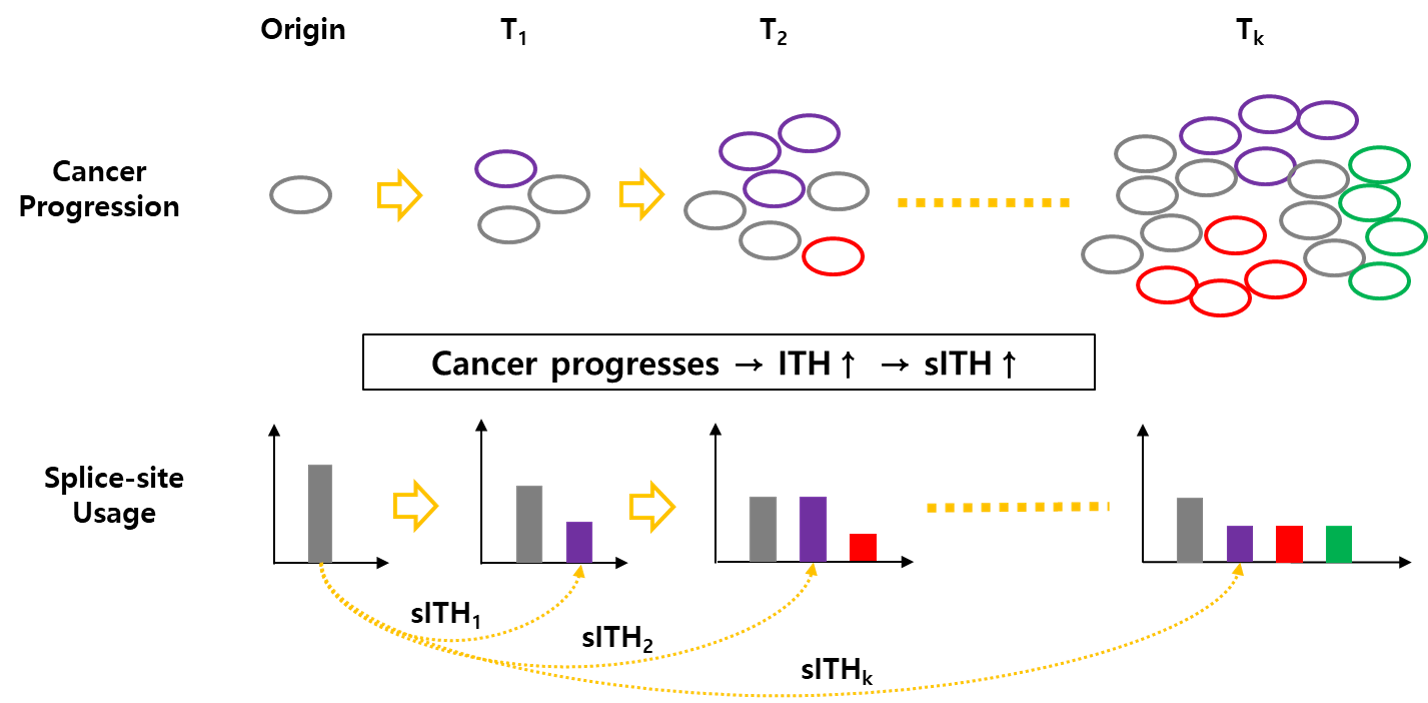
Figure 2. Illustration that describes how the cancer progression affects splice-site usage distribution, which affects spliceomic ITH. Clonal heterogeneity is increased as a result of cancer progression, and the splice-site usage distribution of bulk-tumor changes accordingly. The sITH is also designed to increase accordingly.
SpliceHetero: An information theoretic model for measuring spliceomic intratumor heterogeneity from bulk-tumor RNA-seq
Intratumor heterogeneity (ITH) is the degree of diversity among groups of cells (or clones) that constitute a cancer tissue. The high degree of diversity usually has a negative impact on prognosis by helping cancer tissue acquire malignant phenotypes. Since ITH is a result of clonal evolution, ITH is naturally measured at DNA level. However, ITH at the RNA level can be useful for predicting prognosis as shown in a previous study (i.e. tITH). A natural extension of this study is to measure ITH at spliceome level (i.e, sITH). However, there are some serious technical challenges in measuring sITH from bulk-tumor RNA-seq, such as lack of template network of isforms, and complex splicing patterns. We propose an information theoretic method for measuring spliceomic ITH in cancer cells. The model was extensively tested in experiments with synthetic data, xenograft tumor data, and TCGA pan-cancer data. In the experiments, we showed that the sITH have strong association with cancer progression, clonal heterogeneity, alongwith other important features such as cancer stage, survival outcome, and PAM50 subtype. As far as we know, our model is the first one that defines ITH at spliceome level. The whole process of calculating sITH is organized into a software package and provided for public access.